現在位置 : 貧血 > 貧血與診斷 - Anemia and Diagnosis
|
紅血球平均值的測定,在臨床血液學檢查中很重要,要先測定血比容(Hct)、紅血球數(RBC)、血紅素量(Hb),再計算MCV、MCH、MCHC。
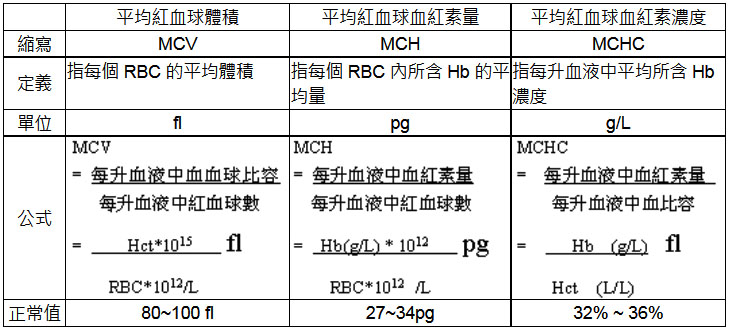
計算 MCV、MCH、MCHC
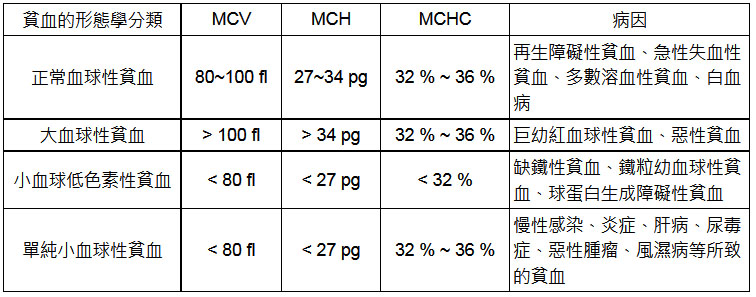
各類貧血的形態學分類
一個重要的概念是網狀紅血球生成指數( RPI , reticulocyte production index ), 正常值為 1 ,計算公式如下:RPI= 網狀紅血球比值× 100/2 × 病患血比容 / 正常人血比容( 45% )
(“ 2 ”是網狀紅血球成熟時間(天),一般網狀紅血球在血液中的生存期為 1 天,若紅血球生成加速,則釋放入血的速度增加,這個是導致網狀紅血球在血液中的生存期則延長至 2~2.5 天。這代表著網狀紅血球數量的增高導致對紅血球生成速率的過高估計。) (貧血診斷)
RDW、MCV與貧血相關性 MCV與RDW的臨床判讀。MCV、MCH、MCHC的定義、正常值範圍,及各類貧血型態學分類。 紅血球體積分佈寬度(red blood cell volume distribution width,RDW)是用血常規分析儀測量獲得的。原理是利用紅血球通過儀器內的計數小孔時,得到相應大小的脈衝訊號,這些訊號經計算機處理後而得到RDW值。因此,RDW是反映外週血紅血球體積大小的指標,一般用RDW的變異係數(RDW-CV)來表示,RDW-CV正常範圍11.5〜14.5%。 臨床RDW用於貧血的形態學分類,或是用於缺鐵性貧血的診斷與鑒別診斷。有時,檢測時並無貧血而RDW超出正常範圍上限,此結果是沒有多大意義的,只能說明紅血球有些大小不均而已。 < MCV與RDW > 以下就MCV與RDW情況將貧血類型及疾病做一說明: 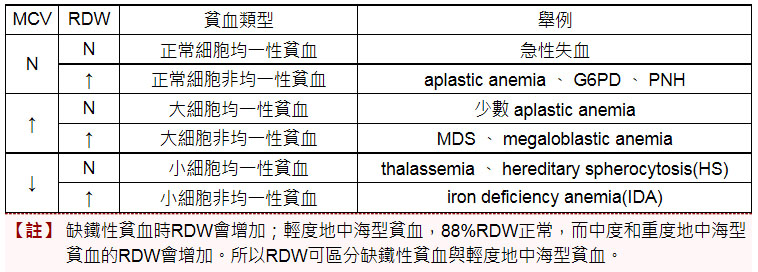
另外,缺鐵性貧血患者在缺鐵潛伏期時RDW就已經增高,治療後以恢復正常無貧血狀態,但RDW仍未降至正常時,可能是體內貯存鐵尚未完全補足。因此,RDW可以用於缺鐵性貧血治療過程的動態監測。
======================================================= |
hemoglobin
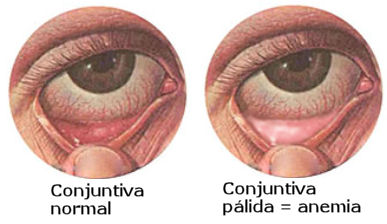
Signs of anemia and normal
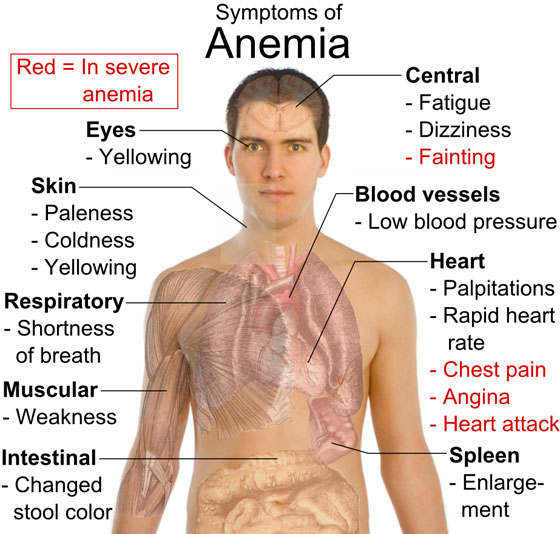
Iron deficiency anemia
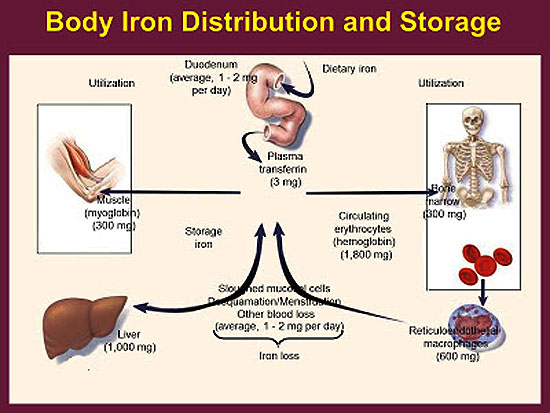
Iron deficiency anemia
|
Click here to edit.
|
再生不良性貧血 (Aplastic Anemia)
再生不良性貧血 (也叫再生障礙性貧血, 簡稱再障貧血),是指骨髓未能生產足夠或新的細胞來補充血液細胞的情況。一般來說,貧血是指低的紅血球統計,但患有再生不良性貧血的病人會在三種血液細胞種類 (紅血球、白血球及血小板)均出現低統計的情況。 病因 現時已知的再生不良性貧血的一個病因是自體免疫性疾病,當中白血球自行攻擊骨髓。很多的病例都不能清楚判斷病因,但再生不良性貧血有時會與一些物質,如苯、輻射的接觸,或是使用某類藥物,包括氯黴素及苯丁吡唑酮有所關聯,可能會造成骨髓造血細胞的器質性病變。 再生障礙性貧血(簡稱再障)(aplastic anemia)是指由各種原因引起的骨髓造血功能衰竭,造成全血細胞減少的一種疾病。臨床上以紅細胞、粒細胞和血小板減少所致的貧血、感染和出血為特徵。再障在中國大陸地區並非少見,中國大陸流行病學調查資料表明其發病率約為0.74/10萬人口,呈散發性。發病以青中年居多,男性略高於女性,原發性稍多於繼發性。 病徵 ● 貧血,有著不適、蒼白及其他病徵; ● 血小板減少症,提高了出血及瘀斑的風險; ● 白血球減少症,提高了感染的風險。 診斷 現時的診斷方法只有骨髓切片檢查。為了尋求診斷的線索,在進行此檢查前,病人一般會接受其他血液測試,包括全面血球統計、腎功能測定、電解質功能測定、肝功能測定、甲狀腺功能測定、維生素B12及葉酸水平等。 治療 再生不良性貧血的治療,可以透過服用藥物或在病症較嚴重時進行骨髓移植。骨髓移植是最能達到痊癒效果的方法,但進行期間卻有很大的風險。從合適捐贈者移植來的骨髓會取代失效的骨髓,骨髓內多能化的幹細胞會重新組成所有血液細胞,建立病患者一套新的免疫系統、紅血球及血小板。但是除了移植失敗外,新造的白血球亦有攻擊身體內其他細胞的可能性,稱為「移植物對抗宿主疾病」。 再生不良性貧血一般治療上開始會採用抗胸腺淋巴細胞球蛋白(ATG或抗淋巴球球蛋白),配合約幾個月環孢菌素的療程,首先壓抑免疫系統,使造血細胞恢復功能,來調節免疫系統。在壓抑免疫系統期間,由於白血球低下,病人會被保護於儘量減少微生物的房間,以減少感染的風險。溫和的化學療法使用環磷醯胺及長春新鹼對治療有效。抗體療法,如使用ATG或目標T細胞卻會攻擊骨髓。使用類固醇一般都沒有效用。 跟進工作 病人需要接受定期的全面血球統計,以確定狀況。 約10-33%的再生不良性貧血的病人會發展出一種罕有的病症,稱為「陣發性夜間血尿症」(簡稱PNH,即貧血以外加上血小板減少症及/或血栓症),一般解釋是指骨髓為對抗免疫系統的攻擊而產生的逃避機制。流式細胞術是一種測試再生不良性貧血的病人是否患上PNH的認可的方法。 Aplastic Anemia
Aplastic anemia (AA) is a disease in which the bone marrow, and the blood stem cells that reside there, are damaged. This causes a deficiency of all three blood cell types (pancytopenia): red blood cells (anemia), white blood cells (leukopenia), and platelets (thrombocytopenia). Aplastic refers to inability of the stem cells to generate the mature blood cells. It is most prevalent in people in their teens and twenties, but is also common among the elderly. It can be caused by exposure to chemicals, drugs, radiation, infection, immune disease, and heredity; in about half the cases, the cause is unknown. The definitive diagnosis is by bone marrow biopsy; normal bone marrow has 30-70% blood stem cells, but in aplastic anemia, these cells are mostly gone and replaced by fat. First line treatment for aplastic anemia consists of immunosuppressive drugs, typically either anti-lymphocyte globulin or anti-thymocyte globulin, combined with corticosteroids and cyclosporine. Hematopoietic stem cell transplantation is also used, especially for patients under 30 years of age with a related, matched marrow donor. Signs and Symptoms Anemia with malaise, pallor and associated symptoms such as palpitations ● Thrombocytopenia (low platelet counts), leading to increased risk of hemorrhage, bruising and petechiae ● Leukopenia (low white blood cell count), leading to increased risk of infection ● Reticulocytopenia (low counts of reticulocytes, that is, immature red blood cells) |
|
Paroxysmal Nocturnal Hemoglobinuria (PNH)
Paroxysmal nocturnal hemoglobinuria (PNH), previously Marchiafava–Micheli syndrome, is a rare, genetically acquired, life-threatening disease of the blood characterized by destruction of red blood cells by the complement system, a part of the body's intrinsic immune system. This destructive process is a result of a defect in the formation of surface proteins on the red blood cell, which normally function to inhibit such immune reactions. Since the complement cascade attacks the red blood cells throughout the circulatory system, the hemolysis is considered an intravascular hemolytic anemia. Other key features of the disease, notably the high incidence of thrombosis, are not totally understood. PNH is the only hemolytic anemia caused by an acquired (rather than inherited) intrinsic defect in the cell membrane (deficiency of glycophosphatidylinositol leading to absence of protective proteins on the membrane). It may develop on its own ("primary PNH") or in the context of other bone marrow disorders such as aplastic anemia ("secondary PNH"). Only a minority (26%) have the telltale red urine in the morning that originally gave the condition its name. Allogeneic bone marrow transplantation is the only curative therapy, but has significant rates of both mortality and ongoing morbidity. The monoclonal antibody eculizumab (Soliris) is effective at reducing the need for blood transfusions, improving quality of life, and reducing the risk of thrombosis. |
|
淺談地中海型貧血 馬偕院訊 2012年8月(320期) ■台北院區兒科門診護理師 曾凌惠 地中海型貧血屬於一種慢性、隱性遺傳溶血性的貧血症,無傳染性,也無法根治,且男女患病機率相同,因好發於地中海沿岸而得名,在中陸長江以南、東南亞和台灣地區也經常可見。為小球性紅血球,低血紅素貧血,是台灣常見的單一基因遺傳疾病之一,故「地中海型貧血」是一種在新生兒就必需要考慮篩檢的遺傳性貧血疾病,其屬於單一基因「自體隱性」的遺傳性疾病,尤以客家籍人較多,目前台灣地區大約有6%的帶因者,其中4%是甲型帶因者,2% 屬乙型帶因者。大部分的病人多屬不會產生任何症狀或對身體健康沒有影響的甲型帶因者,只有極少數病人是屬於乙型帶因者,平均每十三個人中就會有一個帶因者。 何謂地中海型貧血 地中海貧血是一種或多種以上球蛋白多生肽鏈的合成缺陷,且紅血球前驅物質呈現不成熟的破壞。地中海型貧血的分類在於血色素分子中α 和β 鏈異常的差異,而合成α鏈的基因在第16對染色體上共有4個α基因,β鏈的基因在第11對染色體上共有2個β基因,當基因突變缺損造成某種蛋白鏈合成不足時,其他蛋白鏈相對過剩,此種不平衡造成無效紅血球的產生或紅血球體積變小、容易被破壞、壽命縮短、易造成溶血而導致貧血。當α鏈合成不足時稱為α(甲)型海洋性貧血,β鏈合成不足時稱為β(乙)型海洋性貧血。台灣地區以α(甲)型多於β(乙)型。 地中海型貧血的分類 地中海貧血的病人,其血液中血色素的濃度會影響到基因遺傳缺陷程度的輕重,因此臨床症狀以重度、中度、輕度等程度區分。α海洋性貧血輕度患者通常會有輕微貧血但大多無症狀,而重度患者則呈現蒼白且有脾腫大情形。β海洋性貧血輕度患者也會有輕微貧血,亦是無症狀。中度患者會有骨頭畸型、肝脾腫大,偶爾需輸血。而重度患者則有生長遲緩、骨頭畸型、肝脾腫大、黃疸、性腺功能低下,此類型病人因需常輸血,易引起血鐵質沈著導致心肌病變、肝持續腫大、內分泌失調。會因心臟衰竭而死亡,常發生在20~30歲。 診斷此類病人臨床醫師會先尋問家族史、評估臨床症狀、血液常規檢查(CBC、MCV、MCH)、血色素電泳及基因分析。海洋性貧血的篩檢,一般是先以全血計數(CBC)中的MCV小於80 fL或MCH小於25 pg 為認定標準,但是缺鐵性貧血(IDA)與海洋性貧血皆會出現小血球性低血色素貧血,因此在做海洋性貧血DNA診斷時,也應同時檢測ferritin,以確定病人是否合併有缺鐵性貧血。若受檢者ferritin偏低,在補充鐵質後MCV 與Hb 升高成為正常值,且DNA 診斷無海洋性貧血,則應僅為缺鐵性貧血(IDA),否則可能合併患有海洋性貧血。 甲型海洋性貧血為HbA (α2β2)中的α-globin 合成缺陷,而乙型海洋性貧血為HbA 中β-globin合成缺陷,由於此兩種的基因缺損不同,所以事先區分帶因者為甲型或乙型是非常重要的。甲型海洋性貧血帶因者的HbA2 在正常範圍內(<3.5%),大部份的乙型海洋性貧血其HbA2 偏高(>3.5%),但有少數人其HbA2 值正常但是Hb F 偏高,因此HbA2 與Hb F 的定量對區分α型或乙型海洋性貧血扮演重要的角色。為了準確區別帶因者為甲型或乙型,我們不以血色素電泳來測HbA2,因血紅素電泳的錯誤率達10%,而採用高效能液相層析法(HPLC)分析及定量HbA2 和Hb F 值,作為是否進行乙型海洋性貧血分子檢測之依據。但一個人若同時帶有甲型和乙型海洋性貧血的基因,其HbA2 和HbF 上昇的情況會跟乙型海洋性貧血者相同;所以對所有個案皆會進行台灣最常見的甲型東南亞型海洋性貧血的篩檢。 地中海型貧血的治療與防治 輕度的海洋性貧血帶因者,雖不需要治療,但男女雙方考慮結婚生子時,最好接受婚前海洋性貧血的篩檢,及早診斷與防治。在臨床上通常不會產生什麼症狀,所以無法從外表診斷出來,因此除了所有重度海洋性貧血的兄弟姊妹及父母應接受遺傳諮詢外,要預防海洋型貧血的發生,最好的方法就是婚前檢查與產前的篩檢,產前診斷確診率大於90%,懷孕8~12週可做絨毛膜DNA檢查,16~18週可做羊水細胞DNA檢查,18~20週可做胎兒臍帶血DNA及血紅素蛋白分析檢查。 重度的海洋性貧血帶因者需要接受治療包括: (1)輸血治療:由於變形後的紅血球會不斷遭受破壞,所以重型病人必須倚賴終身定期輸血,以維持血紅素的濃度。 (2)排鐵劑治療:輸血的同時亦會輸入過剩的鐵質,這些鐵質到處散佈,沉積到胰臟等各處,造成糖尿病、肝硬化等現象。 (3)骨髓移植:若想要永遠解決重症患者的困擾,唯一的方法就是骨髓移植,不過骨髓移植仍面臨費用昂貴、適當的骨髓捐贈者難覓、感染機率會提高、成功率低的問題。 攝取均衡的營養很重要 地中海型貧血和缺鐵型貧血的發生原因不同,因此千萬不要刻意補充過量含鐵質的營養品或食物,如:牛肉、豬肉、豬血、鴨血、菠菜、蘋果、芭樂、葡萄乾等,反而造成身體上的負擔和傷害。在食物的選擇上和平常人一樣,攝取均衡的營養為最重要,包括豐富充足的蛋白質、尤其是維生素A、B群、C、E及富含葉酸的食物,如:綠色蔬菜、蛋黃、黃豆製品等,均可使身體保持在最佳狀態。 輕度患者無需任何治療,更不會惡化進展為重度患者,千萬不要緊張,平時注意適度運動,但須注意運動傷害,運動可促進新陳代謝,亦可增加血管彈性,有助於輸血時之注射。基於優生保健的原因,婚前記得帶另一半去做檢查,並充分與醫師做最好的討論與諮詢。 日常生活工作後,應有充分的休息及適度的休閒運動,千萬不要熬夜及過度勞累。輕度地中海型患者在生活上及運動上和一般人沒有差異,也沒有特別禁忌。重度患者不宜從事劇烈運動,保持一顆快樂、祥和的心,身心靈將更健康,並應定期與血液專科醫師討論追蹤。 |
